Vizsgálataim tárgya a Ni67Zr33 bázisötvözetre alapuló amorf ötvözetcsoport (fémüveg), melyben a nikkelt cseréltük le különböző mértékben, (Ni67-xZr33Mex) Cu-ra (x=6, 16, 33), Cr-ra (x=5), Pd-ra (x=3). A legtöbb kísérletet Ni67Zr33 összetételű anyagon végeztük. Az ötvözeteket 99,99 % tisztaságú Ni, 99,9 % tisztaságú Zr, 99,999 % tisztaságú Cu, 99,99 % tisztaságú Cr és 99,9 % tisztaságú Pd felhasználásával, vákuumban elektronsugaras olvasztással állítottuk elő. Az öntecseket a megfelelő homogenitás érdekében többször újra átolvasztottuk. Az amorf szalagok védőgáz atmoszférában, olvadék fázisból gyorsan forgó réz hűtőhengerre történő öntéssel (single roll melt spinning) készültek. A kapott szalagok vastagsága 30 ± 5 m m, szélessége 1,3 ± 0,2 mm volt, anyagtól függően, egy szalagon belül a vastagság illetve a szélesség szórása nem haladta meg a 10 %-ot. Az így készült szalagokból ellenállásmérésekhez 10,5 - 11 cm-es, tömegmérésekhez és egyéb vizsgálatokhoz 4-6 centiméteres darabokat használtam föl. Az ellenállásmérések esetében a mérőszakasz hossza rögzítetten 10 cm, a minták felszereléséhez volt szükség hosszabb szalagdarabokra. A felhasznált szalagok felületei nem azonosak, védőgáz atmoszférával érintkező felület pedig fényes, sima, a lövéskor a henger felőli oldalon kialakuló felület matt, érdes (lásd 42. ábra alább).
Előkészítés
A mintákat a vizsgálatokra többféle módszerrel készítettük elő:
Az előkészítés kézzel P400 jelű csiszolópapírba szorítva a 3-szori áthúzással, majd alkoholos tisztítással történt. Ebben az esetbe nem növeltem meg jelentős mértékben a felületet, viszont az előállításkor kialakult felületi oxidréteget mindkét oldalon feltörtem.
A szalagokat meghatározott ideig 4%-os HF oldatban marattam, majd alkoholos papírvattával letöröltem. Ebben az esetben arra számítottunk, hogy a marószer egyenletesen fogja lemarni a felületi oxidréteget, esetleg, mivel a Zr-t jobban oldja a marószer, a felületi nikkel dúsulás jöhet létre. Ezzel a témával a 4.2.4 "A felületi előkészítés, illetve a minta levegővel való érintkezésének hatása a hidrogénfelvétel sebességére" fejezetben foglalkozom.
Egy ellenőrző kísérlet erejéig vizsgáltam olyan mintákat is, melyek felületi állapotát alkoholos tisztításon kívül semmilyen módszerrel nem változtattam meg.
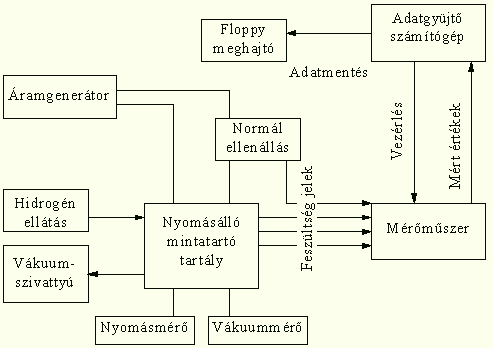
A mintákat - mérésenként 2 darab, egyenként 10 cm hosszú amorf szalag - egy nyomásálló zárt tartályba, elektromosan szigetelt mintatartókra szereljük fel. Egy áramgenerátor segítségével állandó erősségű áramot (50 mA egyenáram) hajtunk keresztül. Mérjük a rajtuk eső feszültséget, és az áramerősség ismeretében kiszámítjuk az ellenállásaikat. A tartályt többszöri H2 átöblítés - vákuumozás (10-3 MPa) után feltöltjük hidrogénnel (0,1-6 MPa). Állandó hőmérsékleten (293-373 K) tartjuk a mintákat és a mintatartókat.
A gáz fázisból történő hidrogén felvételi kísérletek indoklása
Külön kell indokolni, hogy miért gáz fázisú hidrogénnel végeztem a hidrogénezési kísérleteket. Az irodalomban ugyanis szalagok esetében legtöbbször az elektrolitikus feltöltést alkalmazzák. Ezzel a módszerrel nagyobb H/M értékek érhetőek el, lényegesen rövidebb időtartam alatt. A gáz fázisú hidrogénezés mellett azért döntöttünk, mert:
Gáz fázisú hidrogénezés esetén tág határok között, ellenőrizhetően változtatható a hőmérséklet illetve a nyomás. Ezért vizsgálható a telítési H/M nyomás és hőmérsékletfüggése, valamint a hidrid fázis hőmérséklet-stabilitása. Oldatból történő hidrogénezésnél a hőmérséklet változtatása korlátozott.
Gáz fázisból nagyobb mennyiségű minta egyszerre, azonos körülmények között hidrogénezhető, ami igen fontos, ha tömegméréssel akarjuk ellenőrizni a hidrogéntartalmat.
Gáz fázisú hidrogénezés során a hidrogént a mintába hajtó külső erő - az elektrolitikus hidrogén bevitellel szemben - nem csak a minta igen vékony külső felületi rétegére hat, hanem a minta egészére, ezért feltételezhető, hogy a hidrogén eloszlása a mintában homogénebb lesz.
A méréseket egy nyomásálló acéltartályban végezzük, amelyben elektromosan szigetelt mintatartókon helyeztünk el 2 szalagot. A minták végeinél elektromos csatlakozásokat alakítottunk ki, 2-2 csatlakozást az áram be és kivezetésére és 2-2 további csatlakozást, amelyekkel a mintákon eső feszültséget mérjük. Az aktuális áramerősség pontos meghatározásához a körbe beiktattunk egy ismert (és állandó értékű) normálellenállást is. A feszültségjeleket egy nyolccsatornás, számítógépre csatlakoztatható KEITHLEY 199 digitális voltmérő első négy csatornájára vezetjük. A voltmérőt egy számítógép vezéreli, mely a mért feszültség értékekből kiszámítja a minták ellenállását, majd a mérés kezdete óta eltelt idő értékével együtt mágneslemezen eltárolja azt. A mérőrendszer blokk-diagramját a 22. ábra, a gáz hozzá- és elvezető rendszer vázlatát pedig a 23. ábra mutatja be.

A kísérletek során a legtöbb problémát a nagynyomású tartály, s azon belül is a tartály-fedélben elhelyezett elektromos átvezetések okozták.

A feladat azért bonyolult, mert ciklikusan változó nyomás (10-3-9 MPa), változó hőmérséklet (293-373 K, azaz 20-100 °C) mellett kell hidrogénnel szemben gáztömör, elektromosan szigetelt csatlakozást létrehozni. Az első konstrukcióban egy kereskedelmi forgalomban hozzáférhető nagynyomású elektromos átvezetőt alkalmaztunk (24. ábra). Ennél a megoldásnál átvezetőnként 4 db vezeték halad egy kerámiaporral feltöltött, két végén műgyantával lezárt 1,5 mm átmérőjű acélcsövön. A cső kúpmenetes, roppantó-gyűrűs megoldással szerelhető a tartály fedelébe. Ennél a megoldásnál sajnos, mivel a menetnek is tömítenie kell, nehézségek adódtak. Néhány hónapi használat után a menetek mellett - tömítő ragasztó anyag használata esetén is - szivárogni kezdett a hidrogén. Ennek oka feltehetően az, hogy a nyomás-vákuum váltakozása miatt a tartályfedél kis mértékben ugyan, de sokszor oda-vissza deformálódott.
A probléma megoldására egy TDK munka keretében tettünk kísérletet[60]. (25. és 26. ábra) Az új konstrukció teszteléséhez egy próbadarabot készítettünk két átvezetéssel, melyet sikeresen kipróbáltunk.
A gáztér másik tömítendő része a fedél és a tartály csatlakozási felülete volt. Ezt a tömítést sikerült kiválóan megoldanom az összeszereléskor deformálódó élesre kialakított vörösréz tömítőgyűrű alkalmazásával.


Az ellenállásmérés négypontos egyenáramú módszerrel (24. és 27. ábrák), két párhuzamos, független mintán történt.

A mintatartó kialakításánál figyelembe kellett vennünk, hogy a mintákat elektromosan el kellett szigetelni a környezettől, és mindkét minta mindkét véghez 2-2 db (egy áram és egy potenciál) vezetékkel kellett csatlakoznunk. Ezen felül nem volt megengedhető sem a felhasznált anyagok tönkremenetele hidrogén hatására, sem az, hogy vákuumban gázokat szabadítsanak fel. Ezért a mintatartókat kerámia rudakból alakítottuk ki, az elektromos csatlakozáshoz pedig félhenger formájú, a rudakon kialakított fészkekbe illeszkedő vörösréz papucsokat alkalmaztunk. Az elektromos kivezetésekhez nikkel huzalokat használtunk fel, melyeket vékony kerámia, un. pythagoras csövekbe húzva szigeteltünk el egymástól.

28. ábra A mérőprogram folyamatábrája
A mérőprogram megtervezésénél olyan folyamat figyelésére kellett felkészülnünk, amelyben több napig tartó lassú, és néhány perc alatt lezajló gyors változások is előfordulnak. Mivel nem akartunk feleslegesen sok adatot feldolgozni, olyan programot hoztunk létre, mely folyamatosan figyeli a minta ellenállásának változását, de mérési adatot csak 3 feltétel valamelyikének teljesülésekor rögzít. A feltételek a következők voltak:
Az ellenállás az előző öt mérési pontban mért értékek átlagánál egy meghatározott mértéknél jobban eltér.
A legutolsó mérési pont lejegyzése óta egy meghatározott időtartamnál több telt el.
A mérést figyelő személy új mérési pontot kér.
Mindezek alapján a kialakított mérőprogram folyamatábrája a 28. ábrán látható.
Négypontos módszer
Az egyenáramú ellenállás mérésére az irodalomban is igen gyakran alkalmazott un. négypontos ellenállásmérés módszerét alkalmaztuk. A módszer alapváltozatát és az általam alkalmazott kis mértékben módosított változatot az alábbiakban ismertetem.
A legegyszerűbb módszer az ellenállás mérésére az, hogy veszünk egy
áramforrást, egy ampermérőt és egy voltmérőt. Az ampermérőt sorba kapcsoljuk
a mérendő ellenállással és az áramforrással, majd a voltmérőt csatlakoztatjuk
a mérendő ellenállás két végére. Az ampermérőn és a voltmérőn leolvasott
értékekből az Ohm törvény felhasználásával számíthatjuk ki a minta ellenállását.
Ezt a módszert nevezik direkt négypontos ellenállás-mérésnek, ugyanis a
mérendő mintán 4 csatlakozási pontot kell kialakítani. Kettőt az áram be
és kivezetéséhez, kettőt pedig a mintán eső feszültség kivezetéséhez.
Egyszerűsíthető a mérés, ha rendelkezésünkre áll egy ismert értékű
normálellenállás (melynek ellenállás - hőfoktényezője a vizsgálati hőmérséklet
tartomány megfelelően nagy környezetében nulla). Ebben az esetben a normálellenállást
sorba kötjük a vizsgálandó mintával. Az áram nagyságát a normálellenálláson
eső feszültségből számíthatjuk ki (nincs szükség ampermérőre).
A fenti módszer előnyei és hátrányai: a módszer legfőbb előnye egyszerűsége
és kis eszközigénye mellett az, hogy gyorsan, automatizálhatóan szolgáltat
megfelelően pontos ellenállás adatokat. A módszer mérési hibája megnövekszik,
ha a voltmérőn keresztülfolyó áram nagysága egy nagyságrendbe esik a mintán
folyó áram nagyságával. Szerencsére ez a hiba kiküszöbölhető digitális
voltmérő alkalmazásával. Ez a mérési módszer szintén érzékeny a mérőáram
állandóságára. Ha a mérőáram az áram és a feszültségérték leolvasása között
megváltozik, az mérési hibát okoz. Ezt a hibát csökkenthetjük, ha a két
leolvasás között a lehető legkevesebb időt hagyunk.
A minták ellenállását 10 centiméteres mérőhosszon határoztam meg. A mérést teljesen automatizáltam. Az áramerősség értékét minden egyes mérési pont felvételekor egy 10 Ohmos normálellenálláson eső feszültségből számította a program. A mérési pontok felvételekor a számítógép feljegyezte a mérés kezdete óta eltelt időtartamot (percben), majd gyors egymás utáni utasításokkal kapcsolta a nyolccsatornás digitális voltmérőt a normálellenállás, valamint az első és második minta között, bekéri a mért értékeket, elvégzi a szükséges számításokat, és mágneslemezen rögzíti az adott pontban kapott adatokat. Egy mérési ponthoz tartozó feszültség értékek beolvasása 10-15 másodpercet, a számításokkal és az adatok rögzítésével együtt egy mérési pont felvétele 30-35 másodpercet igényelt. Ez a megfigyelt jelenségek sebességéhez tökéletesen megfelelt.
Lehetséges hibaforrások az ellenállásmérés során:
A minták megnyúlásából eredő hiba
A hidrogénfelvétel során a vizsgált amorf minták tágulnak,
nyúlnak. A megnyúlás legnagyobb megfigyelt értéke teljesen telített állapotban
2,5 % volt. Feltételezhető ugyanez az arány a keresztmetszeti irányokban is.
Mivel az ellenállást nem állandó hosszon, hanem állandó végpontok között mérem,
ez hibát okozhat. E hiba nagyságának becslésére az alábbiakban egy mintaszámítást
közlök: (csak a hossznövekedés miatti ellenállás-változást figyelem, itt a fajlagos
ellenállás (r ) változásától eltekintek)
| A minták hossza: | L0 |
| Szélessége: | s |
| Vastagsága: | v |
| Keresztmetszet: | A = s.v |
| Relatív tágulás / megnyúlás: | 1,025 |
| Hidrogénnel telítve a hossz: | LH = 1,025 L0 |
| Hidrogénnel telítve a keresztmetszet: | AH= 1,0252 .A |
A teljes minta ellenállása megnyúlt állapotban, ugyanazokon
a pontokon mérve (ha a fajlagos ellenállás változásától eltekintek) ugyanennyi.
RH=R Számunkra azonban az állandó mérőhosszon mért ellenállás ![]() lenne fontos. Ez a teljes minta ellenállásának 1/1,025-öd része, azaz:
lenne fontos. Ez a teljes minta ellenállásának 1/1,025-öd része, azaz:
RH= 1,0506.![]() ,
ami 5%-os mérési hibát okozhat, ha a fajlagos ellenállást akarjuk kiszámítani
a mért adatokból. Ez a lehetséges hiba két okból is elhanyagolható:
,
ami 5%-os mérési hibát okozhat, ha a fajlagos ellenállást akarjuk kiszámítani
a mért adatokból. Ez a lehetséges hiba két okból is elhanyagolható:
A mérések során kapott ellenállás változások több mint egy nagyságrenddel nagyobbak, mint a megnyúlásból eredő hiba. Ugyanezen véleményen van Y. Yamada és K. Tanaka, akik hasonló ellenállás - hidrogéntartalom méréseket végeztek Ni50Zr50 amorf anyagon[50].
Mérési adataim kiértékelése során nem a fajlagos ellenállás értékeket használom, hanem az állandó érintkezési pontok között mért relatív ellenállás-növekedés időbeli lefutásából vonok le következtetéseket.
A mérési hiba egyik lehetséges oka az, hogy a minták és az elektromos
kivezetések érintkezési pontjaiban termoelemek alakulnak ki, melyek a két
mintán ellentétes irányú hibát okoznak. E hiba nagyságának becsléséhez
mérés közben, 100 °C hőmérsékletnél, az ellenállásértékek jegyzését megszakítva
és az áramgenerátor lekapcsolva, kézi vezérléssel ellenőriztem az érintkezési
pontokon kialakuló parazita feszültségeket. A következő eredményeket kaptam:
| A normálellenálláson (10 ohm) eső feszültség: | Un=0,50280 V | |
| Ebből az áram: | I =50,28 mA | |
| A mintákon eső feszültség: | U1=290,628 mV | U2=311,937 mV |
| A mintákon árammentes állapotban mérhető termikusan gerjesztett feszültség (394 K): | Ut1= - 0,261 mV | Ut2= + 0,166 mV |
| A minták feszültsége és árama alapján számított ellenállás értékek: | R1= 5,7802 W | R2= 6,2040 W |
| A parazitafeszültségek miatti eltérés (hiba): | - 0,00519 W | + 0,0033 W |
| Ez százalékban: | - 0,09 % | + 0,05 % |
A minták ellenállás-hőfoktényezője
További hibaforrás lehetne maguknak a mintáknak az ellenállás-változása a hőmérséklet hatására, azonban a vizsgált anyagok ellenállás hőfoktényezője a vizsgálati tartományban igen kis (a =< 10-4 1/K) érték, (az okozott hiba nagysága 1 % alatt marad), valamint a mérések jelentős része szobahőmérsékleten történt, ahol ez nem okoz hibát.
Szerkezeti változás hosszú időtartamú hőkezelés hatására
További hibaforrás lehet, ha hosszabb hőntartás esetén a mintákban szerkezeti változás zajlik le, amely az ellenállás megváltozásában is jelentkezik. Annak bizonyítására, hogy a hosszú időtartamú 100 °C (373 K) körüli hőntartás nem okoz változásokat az ellenállásban, ellenőrző kísérletet végeztem. 12 db 5 centiméteres, ismert ellenállású Ni51Zr33Cu16 amorf szalag mintát, 6 db vákuum alatt leforrasztott üvegcsőben helyeztem el, majd 100 °C-os kemencébe helyeztem. Ezzel a módszerrel kizártam minden lehetséges kémiai reakciót a hőkezelés során. A mintákat 3, 6, 12, 18, 30 és 42 napos hőkezelés után vettem ki. Az üvegcsöveket lehűlés után feltörve megmértem az ellenállásukat. A kapott értékek 0,1 % hibahatáron belül megegyeztek a hőkezelés előtt mért ellenállásokkal. A fentiek alapján kijelenthető, hogy a hosszú időtartamú 373 K-en végzett hőkezelés önmagában nem okoz a vizsgált anyagokban olyan szerkezeti változást, mely a szalagok ellenállásának megváltozásához vezetne.
Vizsgálataim során a minták hidrogéntartalmának meghatározása során többféle módszer eredményeit is felhasználtam.
1. Tömegmérés
Tömegméréses hidrogéntartalom meghatározáshoz minden esetben egy megtisztított, előre lemért tömegű kvarc kémcsőben helyeztem el 0,6-0,8 g, az ellenállásmérésre használt szalagokkal azonos módon - együtt - előkészített mintát. A kémcsövet a behelyezett szalagokkal együtt ismételten lemértem, majd fölszereltem az ellenállásmérésre használt minták közvetlen közelébe, a nyomásálló tartályba. Ezek után a 2 teljes hidrogénezési ciklust és egy harmadik felhidrogénezést (6MPa, szobahőmérséklet) hajtottam végre. A telítés elérése után az ellenállásmérést felfüggesztettem, a nyomásálló tartányt szétszereltem, majd a tömegmérésre szánt mintákat a kémcsővel együtt ismételten lemértem. A mérés után a kémcsövet visszaszereltem a korábbi helyére, a nyomásálló tartályt lezártam és megkezdtem a minták vákuumozását. Egy tömegmérés rendszerint 15 percet vett igénybe. A vákuumozás közben a fenti folyamatot többször megismételtem, egészen addig, amíg már több órás vákuumozás sem változtatott észrevehető mértékben a minták számítógép által kijelzett ellenállásán. Néhány esetben az utolsó (alacsony H/M-hez tartozó) mérési pontok felvételéhez a vákuumozást kiegészítettem rövid időtartamú felmelegítéssel is, azonban ezekben az esetekben a kályha eltávolítása után mindig kivártam a hőegyensúly beállását.
A feljegyzett tömeg értékekből minden egyes pontban kiszámítottam a minták tömegváltozását, s abból a hidrogéntartalmát. Az egyes pontokban kapott hidrogéntartalom értékeket a tömegmérés előtt és után felvett 2 ellenállás mérési pont relatív ellenállás értékeinek átlagaival (2 minta) párosítottam.
Az alkalmazott mérleg típusa: Mettler M3
Mérhető maximális tömeg: 3 g, a kijelzett tömegérték pontossága 10-6g, de az utolsó számjegy bizonytalan.
Lehetséges hibaforrások: A megszakított deszorpció miatt a minták több alakalommal kerülnek levegővel érintkezésbe. Ezen időszakokban a hidrogén továbbra is áramlik kifelé az anyagból, valamint a felület oxidálódhat. A hidrogén vesztés főleg a telített állapot mérésekor jelentős (itt 6 MPa H2-ből vesszük ki a mintát), azonban még ilyenkor sem figyelhető meg gyors tömegváltozás a mérlegre helyezett mintákon (hiba 10-6g-nál kisebb). Az oxidálódás miatti tömegnövekedés az alacsony hidrogéntartalmú szakaszban okozhat előre nem meghatározható mértékű mérési hibát (addigra gyűlhet föl jelentősebb mennyiségű oxigén a felületen), de itt sem volt megfigyelhető változás az 5 percig a mérlegen tartott mintákon.
A mérleg pontosságából adódó hiba: (mintaszámítás a valóságos tömegekkel)
| A kvarc kémcső tömege | mcs = 1642,837 ± 0,005 mg |
| A kémcső és a minta együttes tömege | m0 = 2240,661 ± 0,005 mg |
| A telitett minták és a kémcső együttes tömege | mtel = 2246,206 ± 0,005 mg |
| Ebből a minta telítetlen tömege | mminta = m0- mcs = 597,824 ± 0,01 mg |
| A hidrogénfelvétel miatt bekövetkezett tömegnövekedés | mH = mtel - m0 = 5,545 ± 0,01 mg |
| A tömegszázalékos hidrogéntartalom | c(wt%) = (mH / mminta ).100=0,9275 ± 0,0017 |
| H/M = (c(t%)/100).MM/MH | H/M = 0,6389± 0,0012 |
A H/M érték fenti hibája 0,2 %-os hibának felel meg. Ugyanezt a számítást a legalacsonyabb mért hidrogéntartalom esetén is végigszámolva a hiba értéke 0,04 %. Ennél nagyobb, de még mindig 1% alatti bizonytalanságot okoznak a korábban felsorolt hibát okozó tényezők.
Itt jegyzem meg, hogy a fenti számítást a méréseim során elért maximális bevitt hidrogén mennyiséggel számoltam végig. A tömegszázalékban megadott hidrogéntartalom ebben az esetben 0,9275 wt% volt.
2. Nukleáris Mágneses Rezonancia (NMR) vizsgálatok
A disszertáció célkitűzéseinek megfelelően az NMR vizsgálatok közül kizárólag csak a hidrogéntartalom meghatározásával foglalkozom. Részletes elméleti fejtegetések nélkül, vázlatosan ismertetem a K. Tompa és munkatársai által alkalmazott vizsgálati módszert, mellyel lehetővé válik több más jellemzővel párhuzamosan a fémüveg minták hidrogéntartalmának meghatározása is.
A magmágneses rezonancia jelensége akkor tapasztalható, ha nem zéró spinnel rendelkező atommagokat külső mágneses térbe helyezünk, majd meghatározott (az adott magokra jellemző) frekvenciájú elektromágneses hullámokból álló impulzus sorozatokkal gerjesztjük. A külső gerjesztés hatására a rendszer lehetséges spin-állapotai között ismert szabályszerűségek szerint átmenetek jönnek létre, melyek során az atommagok energiát vesznek fel az őket körülvevő elektromágneses térből. Impulzus gerjesztés esetében a gerjesztett részecskék igyekeznek visszaállni az eredeti állapotukba. A gerjesztő impulzus megszüntetése után kialakuló válaszjel mérhető megfelelő eszközökkel.
A hidrogént tartalmazó Ni-Zr ötvözetekben a protonok nukleáris mágneses momentuma és 1/2-es spinje a meghatározó. Ehhez viszonyítva a Ni és a Zr magok mágneses momentuma NMR szempontból elhanyagolható.
K. Tompa és szerzőtársai 1996-ban[57] illetve 1997-ben[58] megjelent cikkeikben ismertetik a hidrogéntartalom nukleáris mágnesezettség mérése alapján történő meghatározásának módszerét, melyet az alábbiakban összefoglalok:
A protonok gerjesztéséhez a Carr-Purcell-Meiboom-Gill (CPMG) impulzus sorozatot alkalmaznak, majd mérik a rendszer válaszát (CPMG echo train). A válaszjelnek a gerjesztés kezdőpillanatára extrapolált értéke (M0) arányos az egyensúlyi mágnesezettséggel. Megfelelő impulzus sorozat paraméterek választása esetén a válaszjel amplitúdójának időbeli változása a következőképpen írható le:
A mérési módszer pontosságáról a hidrogéntartalommal arányos hiba nagyságáról és okairól[58] ad részletes ismertetést. Itt csak annyit jegyzek meg, hogy a rögzített "merev" protonok a CPMG impulzus sorozat gerjesztésre 0 választ adnak, ezért ez a mérési módszer a mozgékony protonok számát adja meg amely a vizsgált ötvözeteknél kis mértékben, nagyobb Zr tartalmú ötvözeteknél nagyobb mértékben eltérhet az anyagban oldott hidrogén atomok teljes számától. Az anyagban oldott összes hidrogén biztonságosan kimutatható az alábbiakban ismertettet gáz-kromatográfiás eljárással. A két eljárás által meghatározott hidrogéntartalom értékek közötti különbség megadja az irreverzibilisen (Zr4, esetleg Zr3Ni tetraéderekben) megkötött hidrogén mennyiségét[S7].
3. Gázkromatográfiás mérési módszer
Az értekezésemben közölt, e módszerrel meghatározott hidrogéntartalom értékeket Vasáros L. (ATKI) vezetésével mértem meg. Az általa kifejlesztett módszert és gáz-kromatográfhoz szükséges kiegészítő berendezéseket az ő ismertetése alapján foglalom össze:
Ennél a mérési módszernél a lemért tömegű mintát egy ismert térfogatú kvarccsőben elhelyezett csónakba tesszük. A csónakot, a rendszer argonnal történő átöblítése és lezárása után egy mágnessel a kvarccső egy 1200 °C-ra előfűtött részébe húzzuk. A hő hatására a minták átkristályosodnak és közben hidrogéntartalmuk 81-83 százalékát leadják, mire környezetükkel egyensúlyba kerülnek. Amikor beáll az egyensúly egy mintavevő eszközzel ismert (ált. 1 ml) térfogatú mintát veszünk a gáztérből, amelyet a gáz-kromatográfon keresztül áramló vivőgázba (99,995% tisztaságú argon) fecskendezünk. Ezt háromszor megismételjük. Minden egyes minta előtt egy 99,999 %-os tisztaságú, ismert térfogatú H2 standard befecskendezésével ellenőrizzük a kromatográfot. A kromatográfon belül a 2 m hosszú 4 mm belső átmérőjű kromatográfiás oszlop gázok elválasztására használatos molekulaszűrő anyaggal van feltöltve. Az oszlop hőmérséklete 40 °C-ra, a hővezető-képesség detektoré pedig 150 °C-ra van beállítva. A detektor a hidrogénnek az argonétól jelentősen eltérő hővezető-képessége alapján érzékeli a H2 jelenlétét. A vivőgáz sebessége 15 ml/perc. A detektorból származó jelek megjelennek egy vonalírón, de a kiértékeléshez egy HP integrátor összegzi őket. Az integrátor által megadott értéket a standardra kapott értékkel összevetve, a vivőgáz sebesség ismeretében kiszámítható a beadott gázminta hidrogéntartalma.
Az első három mintavétel után a kvarccsövet gyorsan levákuumozzuk, majd a rendszert lezárva ismét megvárjuk az egyensúly beállását. Ebben a 2. extrakciós szakaszban a minta eredeti hidrogéntartalmának további 11-13 százaléka nyerhető ki. A mintavétel és a mérés az előzőekkel azonos módon zajlik.
Amennyiben a második szakaszban még számottevő mennyiségű hidrogén detektálható, megismételjük. A harmadik szakaszban rendszerint már igen kevés hidrogén detektálható.
A kromatográf által mért, majd integrált jelek alapján kiszámítjuk a kvarccső eredeti hidrogéntartalmát, abból pedig a minta hidrogéntartalmát (táblázatszerkesztő programmal).
Ezzel a módszerrel igen kis mennyiségű hidrogén is nagy pontossággal határozható meg (A számított H/M érték hibája 0,3% alatt van).
4. In-situ tömegmérésen alapuló hidrogéntartalom meghatározási kísérlet
Ennél a módszernél egy nagypontosságú vákuum mérleget alkalmaztunk arra a célra, hogy a deszorpció alatt in-situ mérhessük a minták tömegváltozását és ellenállás változását. Sajnos a mérlegen belül nem alkalmazhattunk fűtést, ezért kis hidrogéntartalmakat nem tudtunk mérni. A másik probléma, hogy a külön berendezésben kétszer ciklizált, majd felhidrogénezett mintákat be kellett szerelni a berendezésbe, majd az egész rendszert levákuumozni mérés előtt. Ezért a teljes telítéshez tartozó pontokat sem tudtuk kimérni.
A mérés a következőképpen folyt: hidrogénezés előtt ismert tömegű kvarccsőben elhelyeztük a tömegmérésre szánt mintákat és fölhelyeztük a mérlegre. Ezzel párhuzamosan egy speciálisan kialakított mintatartóra felszereltünk 1 szálat a minták közül, majd a mintatartót bedugtuk a mérleg vákuumterében kialakított csatlakozóba. 10 mérési pont felvételével rögzítettük a hidrogénezés előtti állapotot.
Ezután a mintatartóra szerelt és a kvarccsőbe helyezett mintákat elhelyeztük egy nagynyomású tartályban, amely rácsatlakozott a vákuum-, és a hidrogénellátó rendszerre, valamint fűthető volt. Ebben a tartályban végeztük el a minták háromszori felhidrogénezését. A harmadik hidrogén-telítés után a mintákat visszaszereltük a vákuum mérlegbe, azt lezártuk, és megkezdtük a vákuumozást. Közben számítógép jegyezte a minták tömegét, ellenállását, hőmérsékletlét, a vákuum mértékét.
Az így kapott adatokból elvileg kisebb korrekciók és számítások után azonnal ábrázolható lenne a hidrogéntartalom - relatív ellenállás-változás görbe, de csak arra a koncentráció tartományra, mely a minták telítése és a szobahőmérsékleten vákuumozással elérhető legkisebb koncentráció között helyezkedik el. Sajnos azonban a technikai jellegű problémák miatt méréseink pontossága megkérdőjelezhető, az így kapott eredmények is fenntartással kezelendők.


